



今日,国家药典委员会发布《凡例》、《微生物限度检查法》等5份国家标准草案,如下:
-
1107微生物限度标准
-
生物制品分包装及贮运管理
-
凡例
-
动物来源药用辅料生产和质量控制指导原则
-
鼠源性病毒检查法(4.荧光定量PCR法)

各标准文件内容如下:
1107 非无菌药品微生物限度标准
非无菌药品的微生物限度标准是基于药品的给药途径和对患者健康潜在的危害以及药品的特殊性而制订的。药品生产、贮存、销售过程中的检验,药用原料、辅料及、中药提取物及中药饮片的检验,新药标准制订,进口药品标准复核,考察药品质量及仲裁等,除另有规定外,其微生物限度均以本标准为依据。
1.制剂通则、品种项下要求无菌的及标示无菌的制剂和原辅料 应符合无菌检查法规定。
2.用于手术、严重烧伤、严重创伤的局部给药制剂 应符合无菌检查法规定。
3.非无菌化学药品制剂、生物制品制剂、不含药材原粉的中药制剂的微生物限度标准见表 1。
表1 非无菌化学药品制剂、生物制品制剂、不含药材原粉的中药制剂的微生物限度标准

4. 非无菌含药材原粉的中药制剂微生物限度标准 表 2。
表 2 非无菌含药材原粉的中药制剂的微生物限度标准
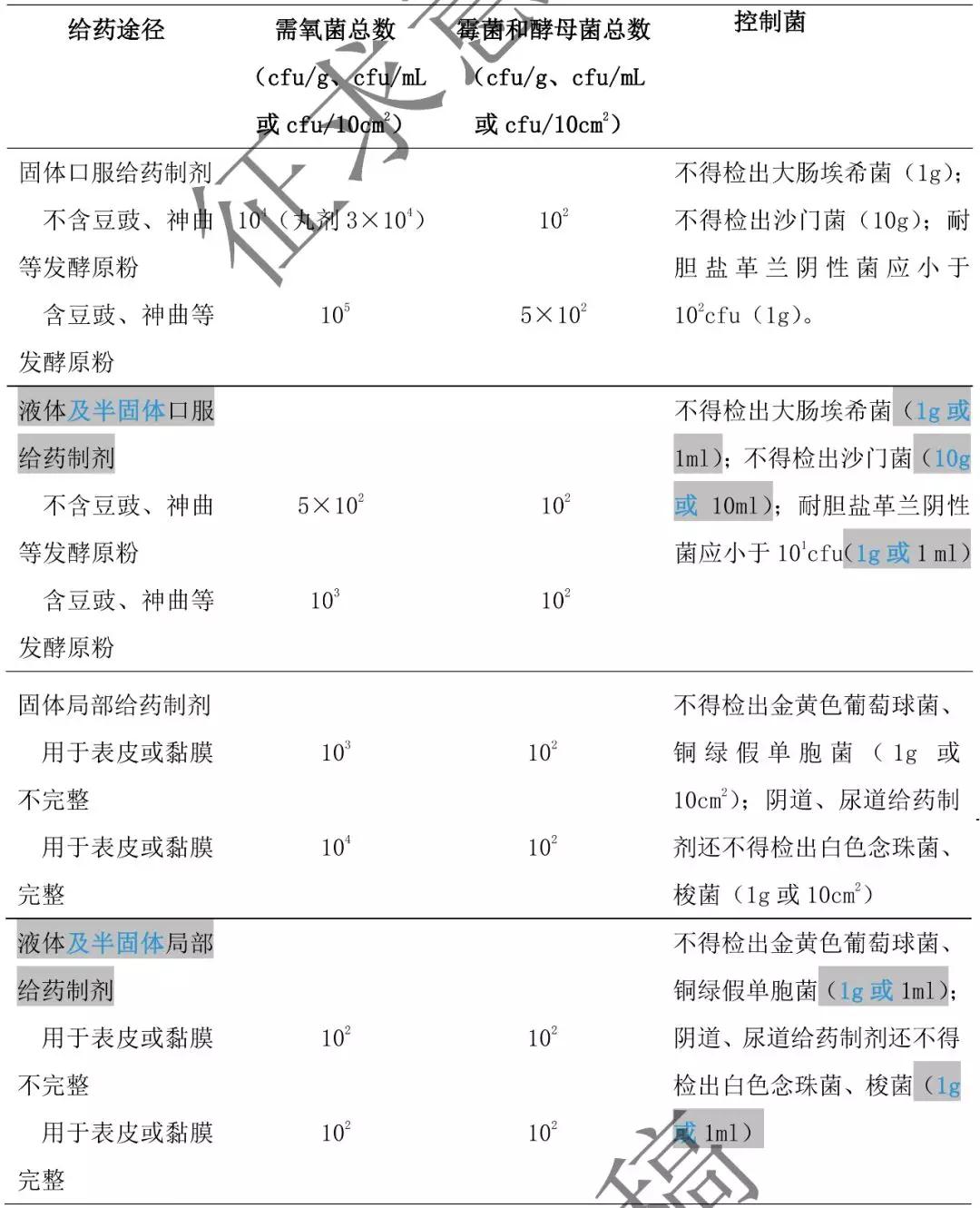
5. 非无菌的药用原料及辅料的微生物限度标准见表 3。
表 3 非无菌药用原料及辅料微生物限度标准
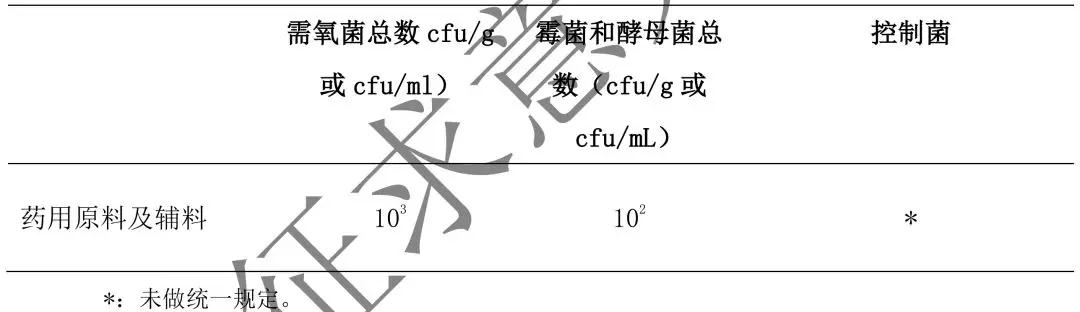
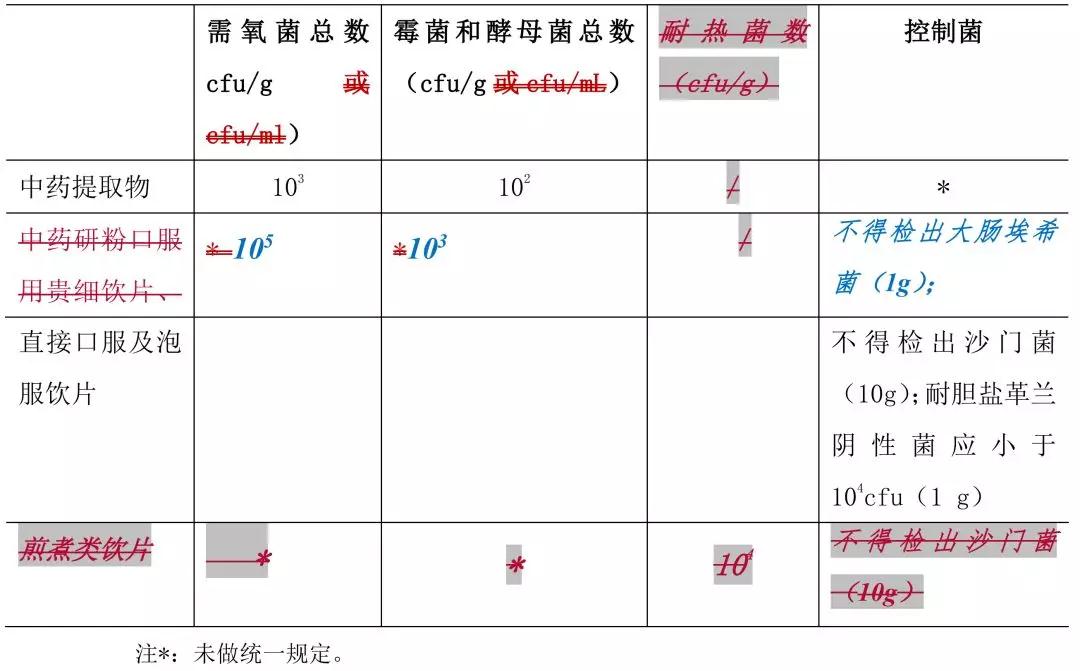
6. 中药提取物及中药饮片的微生物限度标准见表 4。
表 4 中药提取物及中药饮片的微生物限度标准
7.有兼用途径的制剂 应符合各给药途径的标准。
8.除中药饮片外,非无菌药品的需氧菌总数、霉菌和酵母菌总数照“非无菌产品微生物限度检查:微生物计数法(通则 1105)”检查;非无菌药品的控制菌照“非无菌产品微生物限度检查:控制菌检查法(通则 1106)”检查。各品种项下规定的需氧菌总数、霉菌和酵母菌总数标准解释如下:
10^1cfu:可接受的最大菌数为 20;
10^2cfu:可接受的最大菌数为 200;
10^3cfu:可接受的最大菌数为 2000:依此类推。
中药饮片的的需氧菌总数、霉菌和酵母菌总数、耐热菌总数及控制菌检查照“中药饮片微生物限度检查法”(通则 xxx)检查;各品种项下规定的需氧菌总数、霉菌和酵母菌总数及耐热菌数标准解释如下:
10^1cfu:可接受的最大菌数为 50;
10^2cfu:可接受的最大菌数为 500;
10^3cfu:可接受的最大菌数为 5000:
10^4cfu:可接受的最大菌数为 50000:依此类推。
9. 本限度标准所列的控制菌对于控制某些药品的微生物质量可能并不全面,因此,对于原料、辅料及某些特定的制剂,根据原辅料及其制剂的特性和用途、制剂的生产工艺等因素,可能还需检查其他具有潜在危害的微生物。
10. 除了本限度标准所列的控制菌外,药品中若检出其他可能具有潜在危害性的微生物,应从以下方面进行评估:
药品的给药途径:给药途径不同,其危害不同;药品的特性:药品是否促进微生物生长,或者药品是否有足够的抑制微生物生长能力;
药品的使用方法;
用药人群:用药人群不同,如新生儿、婴幼儿及体弱者,风险可能不同;
患者使用免疫抑制剂和甾体类固醇激素等药品的情况;
存在疾病、伤残和器官损伤;等等。
11. 当进行上述相关因素的风险评估时,评估人员应经过微生物学和微生物数据分析等方面的专业知识培训。评估原辅料微生物质量时,应考虑相应制剂的生产工艺、现有的检测技术及原辅料符合该标准的必要性。
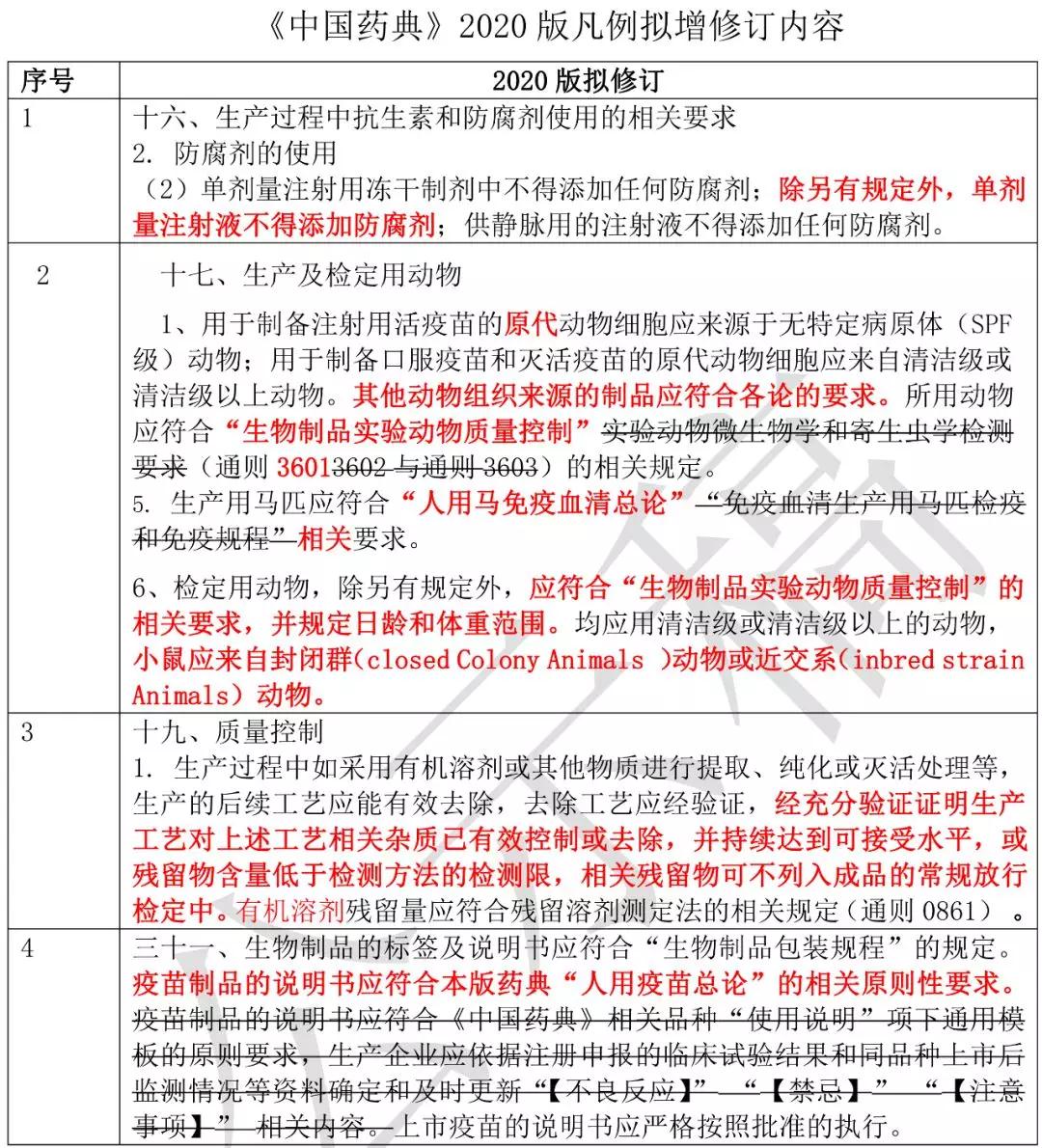
《中国药典》 2020 版凡例拟增修订内容
(见下表)
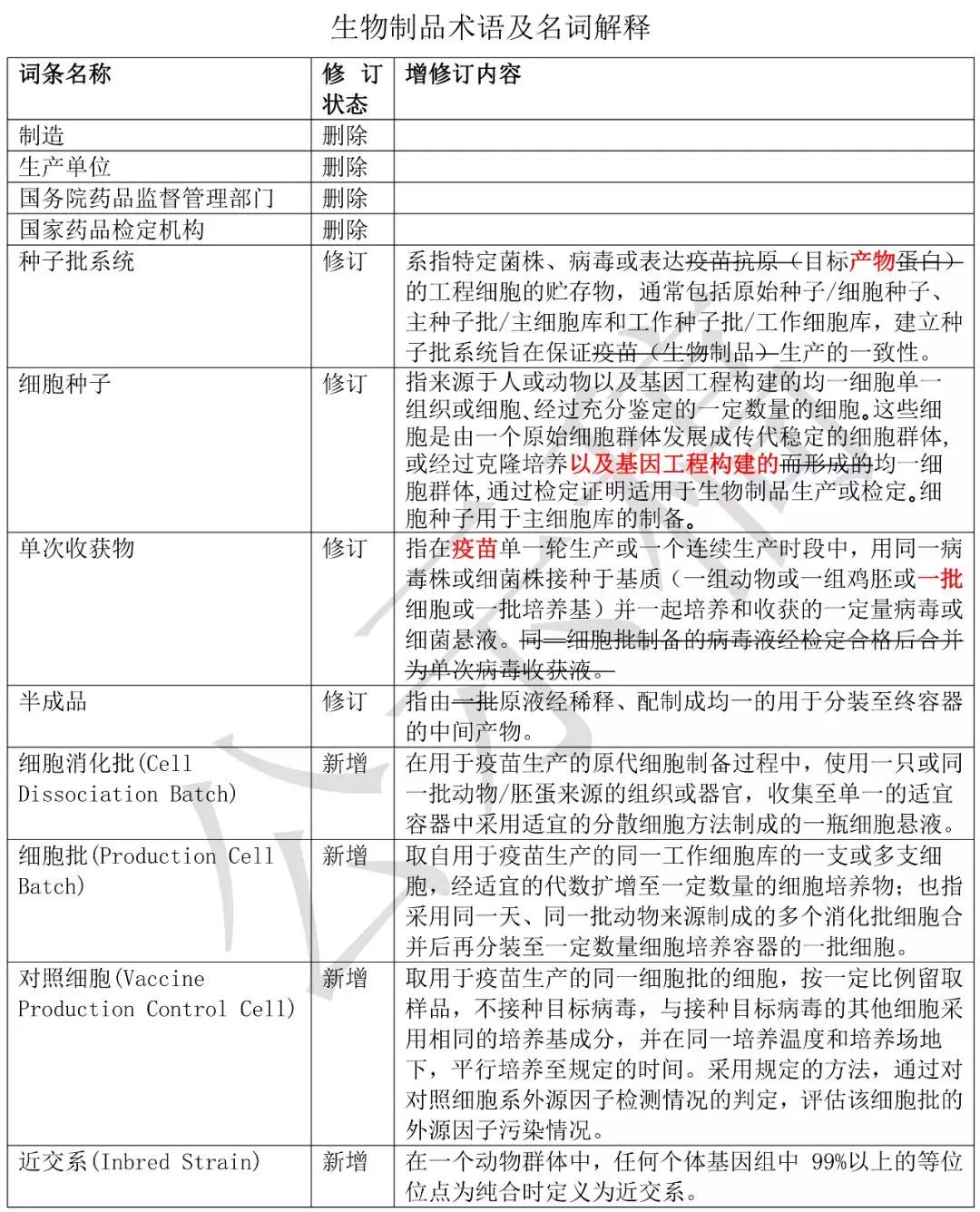
生物制品术语及名词解释
动物来源药用辅料指导原则
动物来源药用辅料系指从动物组织、器官、腺体、血液、体液、分泌物、皮、骨、角、甲等分离提取的,并经充分安全评估的,能够 在药品制剂中添加使用的组分及其加工品。
按原材料来源分类,动物来源药用辅料可分为牛 /羊来源和其他动物来源。按化学组成分类,动物来源药用辅料可分为结构明确的单一化合物(如乳糖)、多种结构明确的单一化合物所组成的混合物(如硬脂酸)、比例和/或结构不明确的多组分混合物(如明胶)等。按工艺制法分类,动物来源药用辅料可分为直接由动物来源原材料制得的分离提取物(如羊毛脂)、分离提取后经过再加工所得的衍生物(如 氢化羊毛脂)等。
动物来源药用辅料通常具有一定特殊性,如原材料的易腐败性、可能存在内源性残留物或外源性污染物(如蛋白、微生物、病毒、农药、兽药等)、组成成分或/和组成比例不明确、特有的对人体有害成分(如朊蛋白)等,从而可能影响辅料质量的批间一致性,甚至引发不可预测的药品不良反应。因此,在药品制剂中添加使用动物来源药用辅料时,应充分评估风险,明确合理性、必要性和可被替代性。
本 指 导 原 则 仅 对直接由动 物 来 源 原材料 分 离 提 取 所 得的 药用 辅料提出相应指导原则,以规范其原材料选择、生产工艺和过程控制、质量研究和稳定性研究、供应商审计等环节的质量控制,以便尽量降低可能存在的风险。本指导原则不涵盖非动物来源原材料制得的药用辅料、由动物来源原材料分离提取后经过再加工所得的药用辅料和人源性药用辅料。本指导原则非强制执行,企业应基于风险管理的理念, 结合药用辅料本身特性及用途开展风险评估及风险防控。
一、原材料的一般要求
动物来源药用辅料的原材料应明确供体动物的入选标准(如健康状况、饲养条件等),一般应固定来源(如牧场、饲养地、屠宰场等)。若发生变更,应重新评估辅料质量及对下游产品的影响。
动物来源药用辅料的原材料一般应保证动物物种或/和种群的同源性,以及取材部位(主要指组织、器官)的一致性。原材料采集后应确定批号,并保证可追溯性。对于已确定批号的原材料,不得擅自 增加批量,并避免交叉污染。
药用辅料生产企业应明确原材料供应商资质,并对原材料质量保证提出控制要求,包括动物脏器提取的操作过程、脏器筛选和收集、 贮藏条件、运输条件等。
作为具有较高风险的原材料, 在使用牛/羊源性原材料进行药用辅料生产和加工时,应提供 TSE/BSE 潜在风险声明。牛/羊的不同组织部位具有不同 TSE/BSE 感染性,根据风险等级,可分为:高感染性组织(如脑、脊髓、视网膜、视神经、脊神经节、三叉神经节、脑下垂体、硬脑膜等)、较低感染性组织(如外周神经、脾脏、淋巴结、食道、胎盘、卵巢、皮肤、肺、肝、肾、血液、奶、尿液等)和无(检出)感染性组织(如睾丸、骨、腱、气管、泪液等)。牛/羊源性原材料一般不得取自高感染性组织;若必须使用,应论证该原材料在辅料生产加工中或相关辅料在制剂生产中的不可替代性,同时原材料的来 源地应符合相关部门的管理规定。
注释:TSE 为 Transmissible spongiform encephalopathies 的首字母缩写,即可传播性海绵体脑炎;BSE 为Bovine Spongiform Encephalopathies 的首字母缩写,即牛海绵状脑病。
二、生产工艺和过程控制
动 物 来 源 药 用 辅料生 产 工 艺 和 生 产过程 的 微 小 变 化 可能 导致 辅料物质基础的变化,从而影响辅料本身及下游产品的质量,因此应严格动物来源药用辅料的变更管理。若生产工艺发生变更,应告知下游 用户,并重新评估辅料质量及对下游产品的影响。
1.生产工艺研究。动物来源药用辅料的生产工艺的开发,应考虑粉碎、提取、纯化、病毒灭活、灭菌等各环节,明确工艺过程的关键环节、控制方法和工艺参数,研究并确定参数范围,经充分验证后,制定操作规程并严格执行,以保证不同批次产品质量的一致性。对环境(如空气、温度等)较为敏感的动物来源药用辅料,生产过程应根据影响辅料质量的外界因素对生产工艺进行控制,如中间体保存条件、 保存时间等。
2.外源因子的灭活/去除。动物来源原材料的残留物及辅料生产过程中引入的污染物,如细菌、真菌、支原体、外源性病毒、农药残留、兽药残留、添加剂及抗生素类药物残留(如三聚氰胺、孔雀石绿)等,可能在辅料使用时引起直接的毒副作用、外源因子污染及有害的免疫应答,引发不良反应。因此,动物来源药用辅料的生产应尽可能采用灭活/去除外源因子的原材料,否则,应在辅料生产过程中增加能够有效灭活/去除外源因子的工艺步骤,明确工艺参数,并对其进行验证,以确保工艺的稳定性和所得终端产品质量的批间一致性,使 灭活/去除的外源因子达到安全水平。
三、质量研究和稳定性研究
动物来源药用辅料的质量控制涉及原材料采集、运输、生产和终端产品贮藏、流通、使用等各个环节,一般应基于风险评估对原材料、中间体、终端产品进行必要的质量研究和稳定性研究,建立全过程质 量可追溯体系。
1.质量研究。质量研究通常可包括理化鉴别、特性分析、功能活性分析、配伍分析等,从而确定相关的分析方法和限度,并建立与制剂特性和给药途径等相匹配的质量标准。对生产过程中的关键中间体,应进行质量研究并建立质量标准。为保证动物来源药用辅料的安全,应对杂质进行深入分析和研究,包括潜在的生物活性杂质;应进行微生物限度检查、控制菌检查和病毒外源因子检查等;对生产过程中可能直接或间接引入的有机溶剂,应保证溶剂残留符合要求;金属元素应有相应控制措施;应确定关键质量属性及控制指标,以评估药用辅 料的批间一致性。
2.贮藏。动物来源药用辅料及其原材料均易受环境影响,一般应选择合适的包材,并进行全面的稳定性研究以确定贮藏条件和有效期/复验期。动物来源药用辅料包装完成后即应置于规定的条件下贮藏。四、供应商审计供应商审计范围和内容应基于风险评估,一般包括对原材料采集和辅料生产、流通、制剂加工等全过程的审计,并保证相关记录可追溯。
对原材料的供应商审计应包括但不限于:动物种属;取材部位;饲养、宰杀、运输过程;贮藏条件;动物检验检疫;外源因子、农药、 兽药残留的风险;供应能力等。
对辅料的供应商审计应包括但不限于:生产资质;生产工艺;工 艺验证;质量控制;贮藏条件和运输过程等。
生物制品分包装及贮运管理
本通用技术要求规定了生物制品生产过程中分批、分装与冻干、包装、贮藏 与运输的具体要求。除另有规定外,均应符合本通用技术要求。
一、分批
批 号系用 以区 分和识 别产 品批的 标志 , 以 避免发 生混 淆和差 错。生物制 品 的批号应由质量管理部门审定。
(一)生物制品批号和亚批号的编制
1. 批 号 的 编 码 顺 序 为 “ 年 月 年流 水 号 ” 。年 号应 写公 历年 号 4 位 数 , 月 份 写 2 位数。年流水号可按生产企业所生产某制品批数编 2 位或 3 位数。某些制品 还可加英文字母或中文,以表示某特定含义。
2. 亚批号的编码顺序为“批号-数字序号”。如某制品批号为:200801001,其 亚批号应表示为:200801001-1,200801001-2,……。
3. 同 一批 号 的 制品 , 应来 源 一致 、 质量 均 一 ,按 规 定 要 求 抽样 检 验后 , 能 对整批制品做出评定。
(二)批、亚批及批号确定的原则
1. 成 品批 号 应 在半 成 品配 制 后确 定 ,半 成 品 配制 日 期 即 为 生产 日 期。非 同 曰或同次配制、混合、稀释、过滤、分装的半成品不得作为一批。
2. 制 品的 批 及 亚批 编 制应 使 整个 工 艺过 程 清 晰并 可 追 溯 , 以最 大 限度 保 证 每批制品被加工处理的过程是一致的,并且是均质的。
3. 单一批号的亚批编制应仅限于以下允许制定亚批的一种情况:
(1) 半成品配制后,在分装至终容器之前,如须分装至中间容器,应按中间 容器划分为不同批或亚批;
(2) 半成品配制后,如采用不同滤器过滤,应按滤器划分为不同批或亚批;(3) 半成品配制后直接分装至终容器时,如采用不同分装机进行分装,应按
分装机划分为不同批或亚批;
(4) 半成品配制后经同一台分装机分装至终容器,采用不同灭菌或灭活设备 进行灭菌或灭活操作、不同冻干机进行冻干,应按冻干机划分为不同亚批 ;同一 亚批制品分装、冻干后,如存在进一步的工艺处理步骤(例如,血液制品分装或冻干 后采用 热处理进行病毒灭活),应基于该 工艺对制品质 量的影响,对每 个处 理单元的制品设置相应的检测项目。
4. 同一制品的批号不得重复;同一制品不同规格不应采用同一批号。
二、分装与冻干
本规 定仅适用 于生物制品的注射剂,其他剂型按“制剂 通则”(通则 0102) 中有关规定执行。
除另有规定外,待分装、冷冻干燥(以下简称冻干)的半成品,须经质量管 理部门确认或批准后,方可进行分装或分装冻干。
(一)分装、冻干用容器及用具
1. 用 于分 装 、 冻干 制 品的 最 终容 器 ,其 质 量 标准 应 符 合 国 家药 品 包装 用 材 料和容器管理的相关要求。应依据制品特性、包材相容性等选择适合的内包材。
2. 分 装容 器 及 用具 的 清洁 、 灭菌 处 理工 艺 应 经验 证 并 确 保 达到 清 洁、 灭 菌 效果。
3. 接触不同制品的分装容器与用具应分别清洗。抗血淸类制品、血液制品、 卡介苗、结核菌素等分装容器与用具必须专用。
(二)分裝、冻干车间及设施
1. 分 装、 冻 干 车间 及 设施 应 符合 现 行中 国 《 葯品 生 产 质 量 管理 规 范》 的 要 求。
2. 分 装、 冻 干 设备 的 规格 和 相关 技 术参 数 应 满足 生 产 工 艺 的要 求 ,设 备 表 面便于清洁消毒,与制品直接接触部件便于拆卸、清洁、灭菌和再利用。
3. 以下情况不得使用同一分装间和分装、冻干设施进行分装、冻干:
(1) 预防类生物制品与治疗类生物制品;
(2) 不同给药途径的疫苗;
(3) 减毒活疫苗与灭活疫苗;
(4) 病毒去除和(或)灭活处理前后与的血液制品。
4. 不 同 品种 及 规格 制品 交 替 使用 同一 分装 间和 分装 、 冻 干 设施 时 应 进 行 共 线使用的风险评估;在一种制品分装后,必须进行有效的清洁和消毒,清洁效果 应定期验证。
(三)人员
1. 直 接参 加 分 装、 冻 干的 人 员, 每 年至 少 应 做 1 次 健 康检 查。凡患 有活 动 性结核、病毒性肝炎或其他可能污染制品的传染病患者,不得参加分装 、冻干工 作。
2. 从 事分 装 、 冻干 的 操作 人 员应 经 严格 培 训 ,考 核 合 格 后 上岗 ;每次 进 入 分装、冻干生产区域的人员应严格限定数量。
(四)待分装半成品的规定
1. 半 成品 自 配 制完 成 至分 装 的放 置 时间 应 经 验证 , 除 另 有 规定 外 ,一 般 应 不超过规定的检定时间。
2. 待 分装 制 品 的标 识 必须 完 整、 明 确, 标 识 内容 至 少 应 包 括品 名 、批 号 、 配制时间、最晚分装时间及分装时限。
3. 存 放待 分 装 制品 的 容器 应 密封 且 无破 损 , 存放 和 转 移 过 程应 采 取严 密 的 防污染措施。
(五)分装要求
1. 分 装设 备 和 无菌 分 装工 艺 应经 验 证;除 菌 过滤 系 统 应 进 行完 整 性测 试 的 验证。
2. 分 装前 应 加 强核 对 ,防 止 错批 或 混批 。分 装规 格 或 制 品 颜色 相 同而 品 名 不同的制品不得在同室同时分装。
3. 分 装过 程 应 严格 按 照无 菌 操作 的 要求 进 行 ,应 进 行 全 过 程的 微 生物 和 悬 浮粒子动态监测并符合要求。
4. 除另有规定外, 制品应尽量采用原容器直接分装,同一容器的制品,应 根据验证结果,规定分装时间,最长不超过 24 小时。
5. 液体制品分装后立即密封,冻干制品分装后应立即冻干。除另有规定外, 应采取减压法或其他适宜的方法进行容器密闭性检查。用减压法时,应避免将安 瓿泡入液体中。经熔封的制品应逐瓶进行容器密封性检查,其他包装容器的密封 性应进行抽样检查。
6. 活 疫苗 及 其 他对 温 度敏 感 的制 品 ,在 分 装 过程 中 制 品 的 温度 应 根据 相 关 验 证试 验 和 稳定性 考察 结果 确定 ,最 高不 得 超过 25°C 。除另 有规 定外, 分装 后 的制品应尽快移入 2〜8℃环境贮存。
7. 混悬状制品或含有吸附剂的制品,在分装过程中应保持混合均匀。
8. 制品实际分装量:
(1)瓶装 液体制 品的实际装量应多于 标签标 示量,应根据所选用 最终容 器 的尺寸,以及待分装制品溶液黏度的不同,适度补加装量,以保证每瓶的抽出量 不 低于 标 签 上 所 标 示 的 数 量 。如 , 分装 100ml 者 可补 加 4.0ml ;分 装 50ml 者 可 补加 1.0ml;分装 20ml 者可补加 0.60ml;分装 10ml 者可补加 0.50ml;分装 5ml 者 可 补 加 0.30ml ;分 装 2ml 者 可 补 加 0.15ml ;分 装 1ml 者可 补 加 0.10ml;分 装 0.5ml 者可补加 0.10ml;
(2)预装式注射器制品的实际装量应不低于标示量。
(六)冻干要求
1. 应 根据 制 品 的不 同 特性 研 究确 定 冻干 工 艺 。冻 干 设 备 及 工艺 应 按实 际 冻 干批量进行验证。冻干过程应有自动扫描记录。冻干全过程应严格无菌操作。
2. 真 空封 口 者 应在 成 品检 定 中测 定 真空 度 。充氮 封 口 应 充 足氮 量 ,氮 气 标 示纯度应不低于 99.99%。
(七)分装、冻干标识和记录
1. 分装、冻干后之制品应有标识,内容至少包括:制品名称、批号、规格、 数量、分装或冻干日期等。
2. 分 装记 录 应 包括 分 装器 具 和过 滤 系统 的 灭 菌处 理 记 录 和 过滤 系 统的 完 整 性测试结果等。
(八)抽样、检定
1. 成 品应 每 批 抽样 进 行全 检 ,如 分 亚批 , 应 根据 亚 批 编 制 的情 况 确定 各 亚 批需分别进行检测的项目。
2. 抽 样应 具 有 代表 性 ,应 在 分装 过 程的 前 、 中、 后 阶 段 或 从冻 干 柜不 同 板 层、不同位点进行抽样;分装过程中如发生可能影响制品质量的偏差时 ,抽样还 应包括对发生上述偏差的适当阶段抽取的样品。
3. 根据实际生产情况,成品检定部分项目可在贴签或包装前抽样进行检定。
三、包装
生 物 制 品 包 装 涉 及 的 说 明 书 及 标 签 管 理 应 符 合 国 家 药 品 监 督 管 理 部 门 的 相 关规定。
(一)包装车间要求
1. 包 装 车间 的 设施 及包 装 材 料应 符合 中国 现行 《药 品 生 产 质量 管 理 规 范 》 要 求。包 装 车 间 应干 净整 洁, 环 境温 度应 不 高于 25 ℃ 。如 制品 贮存 温度 与包 装环境温度不一致,应通过验证确定包装时限。
2. 已分装或冻干后制品,经质量管理部门审核并确认后,方可进行包装。2. 同 一 车间 有 数条 包装 生 产 线同 时进 行包 装时 ,各 包 装 线 之间 应 有 隔 离 设施。外观相似的制品不得在相邻的包装线上包装。每条包装线均应标明正在包装 的制品名称及批号。
(二)灯视检查(以下简称灯检)
1. 除 另有 规 定 外, 熔 封后 的 安瓿 或 密封 的 分 装容 器, 在灯 检 前 应进 行容 器 密封性检查。
2. 制 品在 包 装 前应 按 照各 论 中的 要 求进 行 外 观检 查 , 制 品 灯检 应 符合 以 下 要求:
(1) 人工灯检
① 灯检应采用日光灯(光照度应为 1000〜3000lx),其背景和光照度按制品 的性状调整;
② 灯检人员的视力应每半年检査一次,视力应在 4.9 或 4.9 以上,矫正视力 应在 5.0 或 5.0 以上,无色盲;
③ 凡制 品颜色 或澄明度异常、有异 物或有 摇不散的凝块、有结 晶析出 、封 口不严、有黑头或裂纹等应全部剔除,有专门规定者应按相关各论执行。
(2) 全自动灯检
应对相关设备进行验证,并对比评估全自动灯检和人工灯检的检测效能(如 Knapp-Kushner 测试),设备使用前应进行校准和检查。
(三)标签和药品说明书
1. 药 品包 装 标 签和 说 明书 的 体例 、 规范 和 编 写印 制 应 符 合 《中 华 人民 共 和 国药品管理法》及国家药品监督管理部门的有关规定。
1.药品说明书应与国家药品监督管理部门批准的内容一致。
2.药品包装标签的文字表述应以药品说明书为依据,不得超出说明书内容, 不得加入无关的文字和图案。
4. 应 在说 明 书 中载 明 必要 的 风险 提 示, 以 警 示临 床 使 用 , 如本 品 为皮 内 注 射, 严禁皮 下或肌肉注射(如:皮内注射用卡介苗);人血液制 品应注 明病毒安 全性风险提示供临床使用时权衡利弊。
5. 生 产过 程 使 用抗 生 素、 甲 醛、 裂 解剂 等 原 材料 时 , 应 在 制品 的 说明 书 中注明对所用原材料过敏者不得使用的相关警示语。
(四)包装
1. 已分装或冻干后制品,经质量管理部门审核并确认后,方可进行包装。
2. 包 装 前, 应 按质 量管 理 部 门发 出的 包装 通知 单所 载 明 的 相关 内 容 ( 如 品名、批号、有效期等)准备瓶签或印字戳。瓶签上的字迹应清楚。
3. 包 装过 程 中 应仔 细 核对 相 关信 息 ,防 止 错 误和 混 淆 。在 包装 过 程中 , 如 发现制品的外观异常、容器破漏或有异物者应剔除。
4. 瓶 签应 与 容 器贴 实 ,不 易 脱落 , 瓶签 内 容 不得 用 粘 贴 或 剪贴 的 方式 进 行 修改或补充。直接印字的制品字迹应清楚。
5. 不 同制 品 或 同一 制 品不 同 规格 , 其瓶 签 应 采用 不 同 颜 色 或式 样 ,以 便 于 识别。
6. 每个最小包装盒内均应附有药品说明书。
7. 外 包装 箱 标 签内 容 应直 接 印在 包 装箱 上 。批号 和 有 效 期 采用 打 码机 直 接 打印在包装箱上,字迹应清楚,不易脱落和模糊。
8. 制 品包 装 全 部完 成 后, 应 及时 清 场并 填 写 清场 记 录 , 同 时应 对 包装 材 料 和制品数量进行物料平衡计算;完成包装的成品应及时交送成品库。
四、贮藏与运输
生物制品贮藏和运输管理应符合国家对药品流通和运输的相关要求。本规定适用于生物制品成品的贮藏和运输管理。中间品、原液、半成品的贮藏和运输管理应符合本版药典各论或批准的要求。
(一)生物制品贮藏管理要求
1. 制 品的 贮 藏 条件 ( 包括 温 、湿 度 ,是 否 需 避光 ) 应 经 验 证, 并 符合 相 关 各论或批准的要求,除另有规定外,贮藏温度为 2~8°C。
2. 应 配备 专 用 的冷 藏 设备 或 设施 用 于制 品 贮 藏, 并 按 照 现 行《 药 品生 产 质 量管理规范》的要求划分区域,并分门别类有序存放。
(1) 仓储区的设计和建造应合理。仓储区应当有足够的空间,确保有序贮藏 成品;
(2) 仓储区的贮存条件应符 合制品规定的条件(如温、湿度,避光 )和安全 要求,应配备用于冷藏设备或设施的温度监控系统;
(3) 应对冷库,储运温、湿度监测系统以及冷藏运输的设施或设备进行使用前验证、使用期间的定期验证及停用时间超过规定时限的验证;
(4) 应由专人负责对贮存、运输设施设备进行定期检查、清洁和维护 ,并建 立记录和档案。
3. 应 建 立制 品 出入 库记 录 , 应建 立成 品销 售、 出库 复 核 、 退回 、 运 输 、 不 合格药品处理等相关记录,记录应真实、完整、准确、有效和可追溯。
(二)生物制品运输管理要求
1. 生 物制 品 中 所含 活 性成 分 对温 度 敏感 , 应 在规 定 的 运 输 条件 下 采用 最 快 速的运输方式,缩短运输时间.
2. 除 另 有 规定 外, 应 采用 冷链 运 输 。冷 链 运输 ,即 运输 全 过 程, 包括 装 卸 搬运、转换运输方式,外包装箱组装与拆除等环节,都能使制品始终保持在一定 温度下。疫苗冷链运输应符合国家相关规定。
3. 采 用冷 链 运 输时 , 应对 冷 链运 输 设施 或 设 备进 行 验 证 , 并定 期 进行 再 验 证;应由专人负责对冷链运输设施设备进行定期检查、清洁和维护,并建立记录 和档案。
4. 制 品的 运 输 温度 应 符合 各 论或 批 准的 温 度 要求 , 温 度 范 围的 确 定应 依 据 制品的稳定性试验的验证结果。
5. 运输时应避免运输过程中震动对制品质量的影响。
6. 生 物制 品 运 输过 程 中可 能 存在 难 以避 免 的 短暂 脱 冷 链 时 间, 应 依据 脱 冷 链时间和温度对制品质量影响的相关研究,确定可允许的脱冷链时间和可接受的 温度限度。